Highlights
Below you find highlights of the Papp group.
See R. Streber et al. Angew. Chem. Int. Ed. 48 (2009) 9743,
or our review: C. Papp et al. Surf. Sci. Rep. 68 (2013) 446.
Oxidation of sulfur on flat and stepped platinum surfaces
That’s how the reaction of sulfur with oxygen looks like
in a TP-XPS experiment
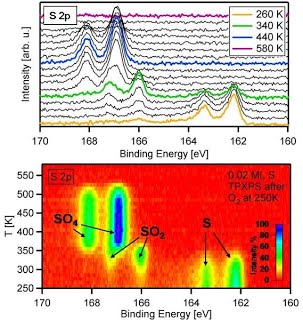
and its analysis
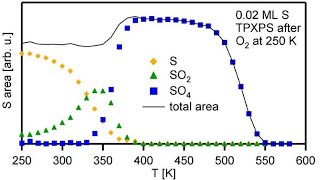
and in isothermal experiments:
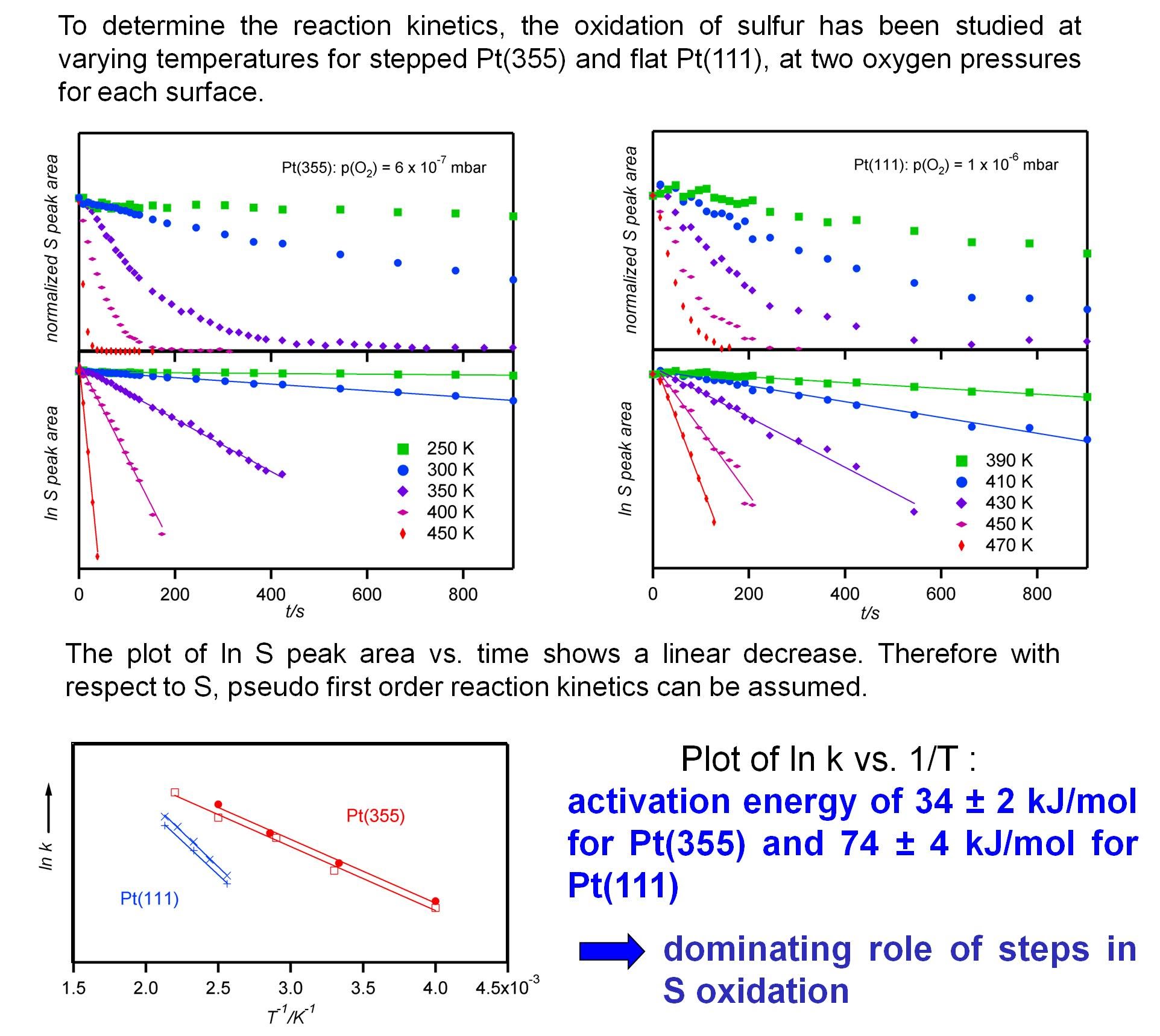
The results show us the reaction enthalpy of the rate determining step, show the role of steps in this reaction.
Please also see our review: C. Papp et al. Chem. Rec. (2014) DOI: 10.1002/tcr.201402014.

In our research we aim at the understanding of the dehydrogenation mechanisms of the LOHC material perhydro-N-ethylcarbazole on the model catalyst Pt(111).

From the XPS data shown above we gained detailed insight to the reaction intermediates and reaction temperatures.

This work is funded by BMW and the ‘Cluster of Excellence Engineering of Advanced Materials’ at the University Erlangen-Nürnberg. The work is published in the journal ChemSusChem together with our colleagues from physical chemistry (J. Libuda; website) and from chemical reaction engineering (P.Wasserscheid; website).
In situ XPS during the Growth of Graphene on Ni(111)

Surface Structures of Graphene on Ni(111)

The graphene layers formed on Ni(111) exhibit two differrent, energetically almost equivalent structures, bridge-top and top-fcc.
This work was published in J. Phys. Chem. Lett. together with our colleagues from the institute of theoretical chemistry in the working group of A. Görling (website).
Doping of Graphene on Ni(111)
We doped graphene with nitrogen using two different methods:

low energy nitrogen ion implantation and growth of graphene with a nitrogen containing precursor.
To analyze the changes in the band structure due to doping we used ARPES.

The DFT analysis showed a dependence of the doping (n and p doping) on the bonding geometry of nitrogen in graphene.

This work is published in Phys Rev B together with the groups of Th, Seyller (website) and A. Görling (website)
Carbon Dioxide Capture in Ionic Liquids
In this study we investigated the reaction mechanism for CO2 capture reaction of a ionic liquid in the outermost layers and compared them to its bulk. We were able to give a detailed analysis of the reaction products and the differences in the near surface region compared to the bulk IL from our NAP XPS measurements.
This study provides a fundamental insight into the reactions that occure in Carbon Capture and Storage (CCS) applications.
This study was conducted in collaboration with the group of F. Mayer (PC II) and P. Wasserscheid (CRT), Uni Erlangen. For more information please see I. Niedermayer et al. J. Am. Chem. Soc. 136 (2014) 436.


Interaction of Ethanol with Co/CeO2(111)

The adsorption of ethanol on ceria and cobalt particles on ceria were investigated at pressures of up to 1 mbar. The adsorption of ethanol on the clean ceria lead to a gradual reduction with increasing pressure already at 300 K, with the reduction being more pronounced at higher temperatures. The main intermediate, ethoxide, was formed by dissociative adsorption of ethanol at room temperature. Upon deposition of metallic cobalt on ceria, partial reduction of ceria and the oxidation of the cobalt particles to Co 2+ was observed. The reaction of ethanol with the Co/CeO2(111) model catalyst up to 600 K led to severe reduction of cobalt and ceria, as well as coke formation.
This study was a collaboration with the University of Szeged, Hungary. Please see L. Ovari, et al. J. Cat. 307 (2013) 132 for more information.
The idea of these studies is to study a highly defined system that allows to investigate the fundamental surface chemistry on metal clusters. On lattice mismatched substrates, the spatially varying interaction strengths between graphene and the substrate leads to a Moiré pattern.Such corrugated graphene sheets are outstanding templates for the fabrication of nanocluster arrays, with a narrow size distribution of the clusters.
To understand catalytic processes on the atomic level, it is necessary to find suitable model systems. An appropriate model system must be simple enough to analyze it in detail yet complex enough to accurately reproduce the features of a real catalyst, which is given on for these nanocluster arrays.

On these clusters we determined the adsorption sites, site occupation and site preferences in situ in a quantitative way, to elucidate the adsorption properties. Also poisoning of the catalysts by e.g. by sulfur can be investigated in details. Even the reaction of carbon monoxide with oxygen is accessible, yielding significantly lower activation energies than for the flat single crystal surfaces.
These data become possible due to a small size distribution of the homogeneous cluster arrays, as seen in typical STM data from Pt clusters on h-BN / Rh(111). The h-BN Moire serves as a template and leads to well separated clusters that are stable even at reaction temperatures.

Typical data of such a reaction experiment is shown below, for the isothermal reaction of adsorbed oxygen with carbon monoxide.

It turns out that in comparison to the single crystalline surfaces the reaction order changes from a ~0.5 order due tot he reaction at the edges of islands to a pseudo first order reaction as no extended islands are formed on the clusters. The reaction rate is higher with an activation energy of only ~13 kJ/mol.
Further reading: